Powered By


Saccharomyces cerevisiae has long been the most tractable organism for eukaryotic cell biology, owing to its genetic malleability, greatly facilitated by a preference for homologous recombination (HR) over non-homologous end joining (NHEJ) for double stranded break (DSB) repair. Over the years, biologists have taken advantage of this preference, allowing for the site-specific installation of genetic material and genomic edits with base-pair precision (Moqtaderi and Geisberg, 2013).
However, the demands on yeast engineering have significantly increased with the design of more complex systems and extensive metabolic pathways. Genetic techniques that have historically relied on marker recycling are proving too cumbersome to keep up with the ambitions of synthetic biologists.
Since the initial reports of CRISPR/Cas9 mediated genome editing in yeast by Dicarlo et al. in 2013, a diverse number of novel strategies have been developed for Cas9 and guide RNA expression, allowing highly efficient genome editing that can be multiplexed without the need for a marker (see Jakočiūnas et al., 2016 for a review on these).
Two of these new strategies are particularly powerful, improving the structure of the sgRNA - allowing for high expression and stability of the sgRNA in yeast (Ryan et al., 2014), as well as the method of sgRNA delivery - selecting for cells proficient in gap repair (Horwitz et al., 2015). Combined, these two advancements allow for high-efficiency, markerless editing of the yeast genome, aiding the work flow within the Ellis Lab.
CRISPR/Cas9 can be used for a number of strain engineering purposes, including:
The greatest impact of CRISPR/Cas9 to the daily workflow of the yeast synthetic biologist is likely to be the integration of multiple expression cassettes into the genome without the need for a marker. Chromosomal integration has several benefits over vector based maintenance of foreign DNA:
Fig 1 highlights the importance of genomic integration for stable, robust and repeatable expression. Although plasmid toolkits allow for the routine integration of genetic cassettes into the yeast genome, there is still a need to identify colonies that have integrated the exogenous DNA using a selectable marker. As markers are limited, even in laboratory auxotrophic strains, they can quickly become used up as a project becomes more complex, limiting strain development further down the line.

CRISPR/Cas9 avoids this complication as the integration of DNA into the yeast genome has become so efficient that selection is no longer a necessity, rather, a small handful of colonies can be screened using a simple colony PCR to validate successful integration. Furthermore, integration can be multiplexed, and is amenable to randomized library approaches (Horwitz et al., 2015), permitting a plethora of permutations to be assembled within a single transformation. For this reason, CRISPR/Cas9 and markerless integration of expression cassettes should be considered as a standard for introducing genes of interest into yeast, and a number of parts have been generated in the Ellis Lab to facilitate this (see parts list).
A recent advancement in yeast CRISPR/Cas9 technology was the development of highly persistent intracellular sgRNAs via the fusion of a 5’ HDV ribozyme and a yeast tRNA (Ryan et al., 2014). The addition of the tRNA allows the use of a host species specific promoter, which has been one of the limiting factors in achieving high levels of sgRNA expression in a number of organisms.
The ribozyme was included to serve serve two purposes:
Combined, these two structures improved the efficiency of gRNAs enough to make multiplexed CRISPR/Cas9 genome editing possible without the need for selection.

Another important development in yeast CRISPR/Cas9 was the novel delivery of sgRNA into yeast cells, by introducing the vector as linear DNA fragments that have to be assembled by the yeast HR machinery to reconstitute the sgRNA expression vector (Fig. 3). Horwitz et al. (2015) proposed that this ‘gap repair identifies a special subpopulation of transformed cells’:
'Competency for homologous recombination is known to vary throughout the cell cycle, and the asynchronous mixture of log-phase cells used for transformation would be expected to contain cells in all states. If yeast cells most proficient in gap repair are also best equipped to accomplish multiplex integrations, then this capability may allow markerless, multiplex integrations to proceed with only transient expression of the sgRNAs.'
Although it must be noted that this method requires initial integration of Cas9 into the genome, under a medium/weak promoter.

The Ellis Lab Cas9 and sgRNA expression tools were developed using the Yeast MoClo Toolkit (YTK), produced by Lee et al. (2015) at UC Berkeley, along with additional parts created by ourselves. These new tools combine the highly efficient sgRNA structure, yeast optimised Cas9 ORF, and the novel sgRNA delivery based on the gap repair of linearized fragments (Ryan et al., 2014; Horwitz et al., 2015). Furthermore, the Cas9 ORF was incorporated into the sgRNA expression vector so it may be introduced into the cell at the same time, removing the necessity of constitutive expression of Cas9 from the genome.
The toolkit consists of:

The sgRNA vector is completed using small fragment Golden Gate assembly (see protocols), which is digested with EcoRV to generate a linearized expression cassette. This linearized cassette has 500 bp arms with exact homology to the Cas9-sgRNA expression vector, and will gap repair the the linearized preparation of this plasmid in yeast to to reconstitute a stable Cas9-sgRNA expression vector (Fig. 5b). The marker on the Cas9-sgRNA vector can be used to select for successfully transformed cells expressing both Cas9 and an sgRNA.

As the sgRNA cassette and Cas9-sgRNA gap repair vector are always in the same format, once the sgRNA cassette has been linearized it can be used with any Cas9-sgRNA gap repair vector, and in combination with any other linearized sgRNA cassette, allowing for multiplexed Cas9 targeting (Fig. 5). Although only one linearized sgRNA cassette gap repairs the Cas9-sgRNA expression vector, it seems that expression from the linear pool of intracellular sgRNAs is sufficient, and editing is likely to occur early on within the first mother cell. We have successfully used up to 8 sgRNAs within a single transformation.
Although incredibly powerful, multiplexed CRISPR/Cas9 has its limitations. Selecting more than 3-4 targets to edit on the yeast genome will begin to stretch the capabilities of markerless editing beyond what is a reasonable number of transformants to screen. Despite advances we have made towards in silico prediction of gRNA ‘on-target scores’, in reality, efficiencies will be influenced by other factors, such as local chromatin structure. This makes it incredibly hard to forecast how successful any particular multiplexed edit will perform.
To address the limitations of CRISPR/Cas9 multiplexed editing, we explored other solutions to speed up cell line development requiring a large number of modifications. We identified 3 markers that can be used to iteratively perform edits without the need to cure the cells of the previous CRISPR/Cas9 machinery, by simply transforming the cells with plasmids containing the next marker in the cycle (Fig. 6).
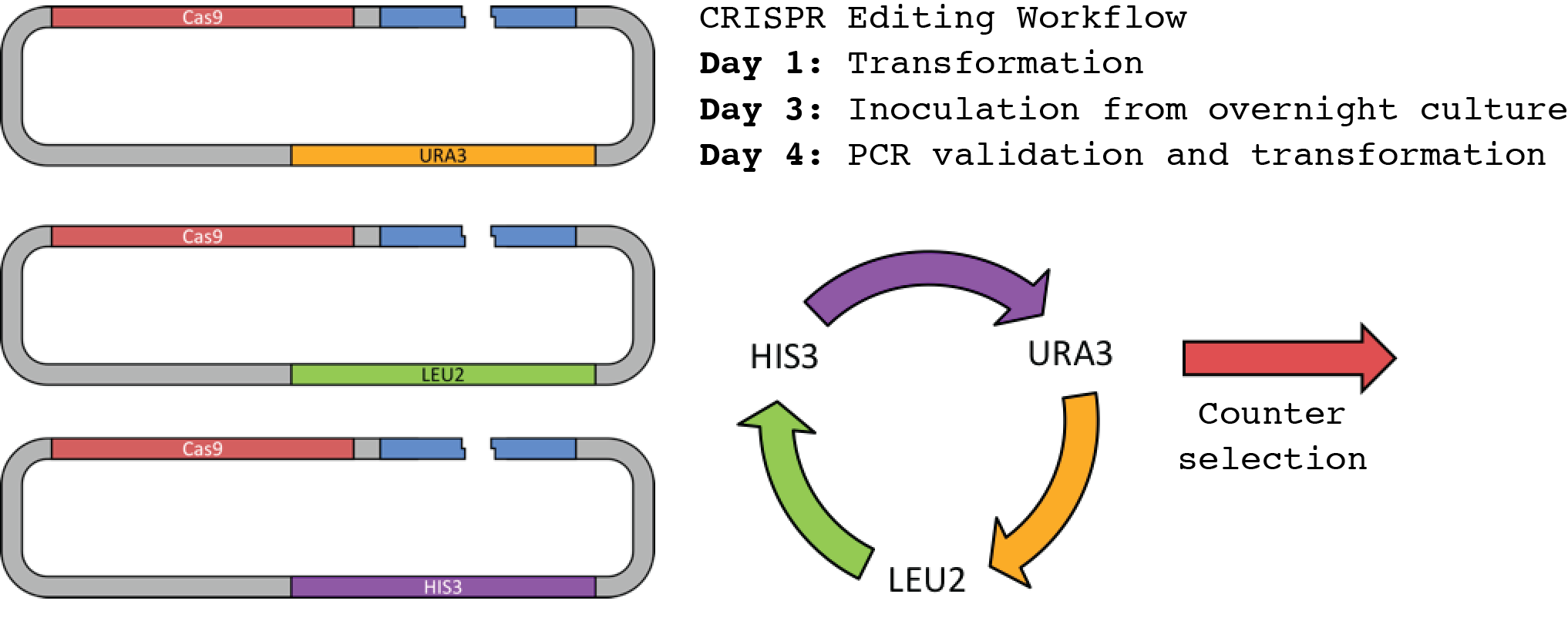
A single rotation around the cycle is sufficient to arrive at zero background from the initial marker. i.e. If you start by using the URA3 marker, once you return to the URA3 marker, you will not get any growth from untransformed cells on a uracil deficient plate.
This makes it possible to immediately transform cells, once they have been validated by colony PCR, reducing the time between edits by several days. Using this workflow, and multiplexing 3-4 edits per transformation, it is possible to achieve 9-12 edits within 2 weeks.
| Name | sgRNA promoter | Yeast marker | E.coli marker |
| pWS082 | tRNAPhe | N/A | AmpR |
| Name | Cas9 promoter | Yeast marker | E.coli marker |
| pWS158 | pPGK1 | Ura3 | KanR |
| pWS171 | pPGK1 | Leu2 | KanR |
| pWS172 | pPGK1 | His3 | KanR |
| pWS173 | pPGK1 | KanR | KanR |
| pWS174 | pPGK1 | NatR | KanR |
| pWS175 | pPGK1 | HygR | KanR |
| pWS176 | pPGK1 | ZeoR | KanR |
| pWS160 | pRPL18B | Ura3 | KanR |
| pWS177 | pRPL18B | Leu2 | KanR |
| pWS178 | pRPL18B | His3 | KanR |
| pWS179 | pRPL18B | KanR | KanR |
| pWS180 | pRPL18B | NatR | KanR |
| pWS181 | pRPL18B | HygR | KanR |
| pWS182 | pRPL18B | ZeoR | KanR |
| Name | Integration loci | Yeast marker | E.coli marker |
| pWS471 | Ura3 | N/A | KanR |
| pWS472 | Leu2 | N/A | KanR |
| pWS473 | HO | N/A | KanR |
| Name | Integration loci | Yeast marker | E.coli marker |
| gWS044 | Ura3 | N/A | AmpR |
| gWS042 | Leu2 (1/2) | N/A | AmpR |
| gWS043 | Leu2 (2/2) | N/A | AmpR |
| gWS045 | HO | N/A | AmpR |
To design and analyse your guide RNAs please refer to Designing and Analysing Guide RNAs on the tutorial page of Benchling for an overview of how to choose your guide RNA in a particular region of the yeast genome. You can then refer to sgRNA Small Fragment Assembly for a protocol on assembling your two short oligos into the sgRNA entry vector (pWS082). Once assembled and sequence verified, the sgRNA expression cassette needs to be linearized with EcoRV. sgRNAs can be pooled with other sgRNAs and digested together. The entire mixture is then added to the remaining CRISPR components (linearized Cas9-sgRNA gap repair vector and donor DNA) and transformed directly into yeast. See sgRNA preparation for details on digestion conditions.
Before using any Cas9-sgRNA gap repair vector it should be linearized using BsmBI and gel purified. It is easiest to create a linearized stock of each vector by digesting and purifying an entire miniprep of the plasmid and setting the concentration to 100 ng/ml. Each transformation requires 100 ng of the gap repair vector, so should last a long time.
Markerless cassettes have been created to be fully compatible with the YTK hierarchical workflow, and once complete, can be integrated with the aid of the CRISPR/Cas9 machinery (gRNAs for targeting the markerless integration cassettes also in the parts list. Before transforming, integration cassettes require digestion with NotI (see final part of Yeast Transformation – High efficiency for digestion conditions). For more information on how to use the YTK toolkit, see supplementary information in Lee et al.
For transformation of CRISPR components, a high-efficiency transformation protocol should be used. The Yeast Transformation – High efficiency protocol is sufficient for routine CRISPR/Cas9 experiments, however, for multiplex experiments with 3 or more edits, it is recommended you use Yeast Transformation – Super-High efficiency.
For genome editing you will need 3 DNA components:
These are rough guidelines that seem to work pretty well. The linearized Cas9-gRNA expression vector and sgRNA seem to work well at these amounts, however the amount of donor DNA will vary widely with your particular application and design – this will be down to your best judgment, however, larger amount of donor DNA seem to overcome efficiency problems. This can require anything up to 5 µg DNA of each donor.